Bioinformatic tool: Read_extractor
Agustín Sabbione
Read_extractor 🧬📊
Developed during my PhD, Read_extractor is a Linux script crafted to efficiently extract reads from specified genomic loci in a sorted and indexed BAM file, utilizing Samtools. It reads loci from a .csv file, retrieves reads from each locus, counts repetitions, and adds counts in a new column.
Usage
-i: Path to input file. Must be a sorted and indexed BAM file.
-f: Path to csv file containing the list of loci to extract. You can easily format your loci to csv using Sed, e.g: if you have your loci in a calc or excel sheet, you can change the new line (
\n) to comma (,) with sed ':a;N;$!ba;s/\n/,/g' inputfile > output.csv .-s: Select sense or antisense strand. Works with SAM flags, select 0 for sense or 16 for antisense.
-r: Minimun number of reads to keep.
Example
./Read_extractor.sh -i input_sorted.bam -f Loci.csv -s 0 > ./output/sense.tabUsing
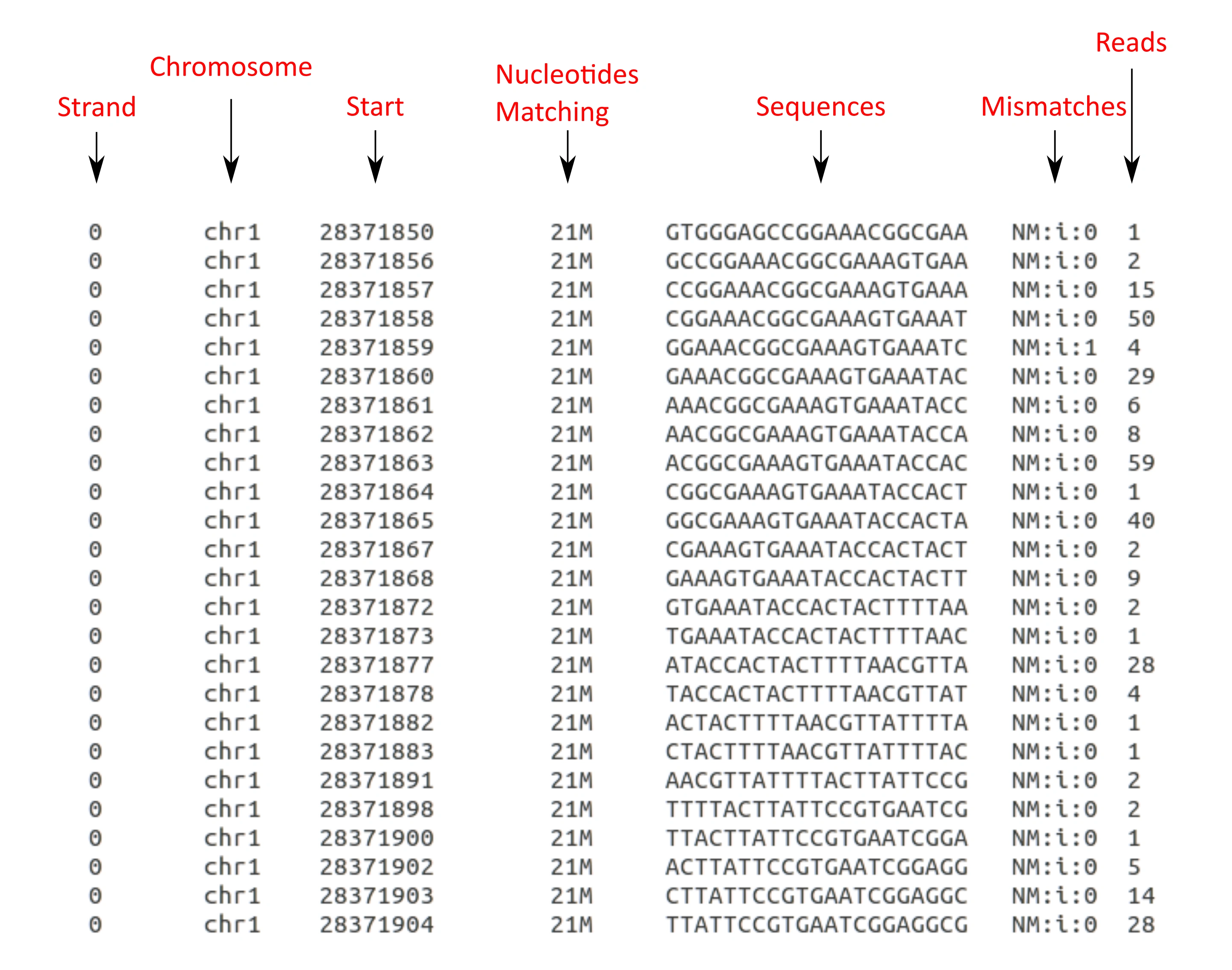
Read_extractor_individual.sh will generate one file for each locus in the loci file, it will put the output files in the working directory, don't add ">".Your output should look like this one:
Like this project
Posted Feb 7, 2024
Simple Bioinformatic Linux script to obtain reads from several genomic loci in a sorted and indexed BAM file
Likes
0
Views
12
Tags